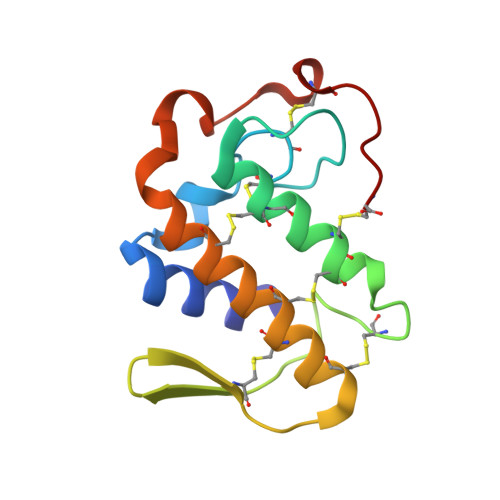
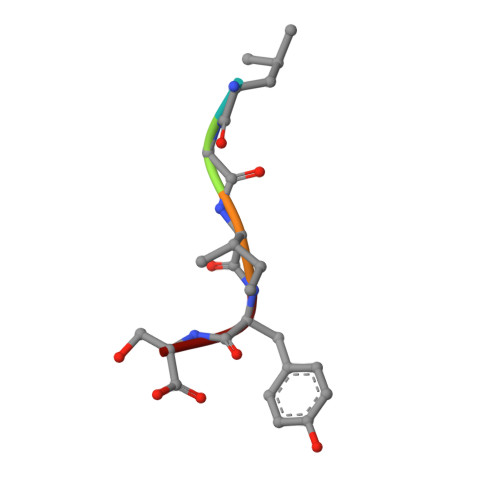
Design of specific peptide inhibitors of phospholipase A2: structure of a complex formed between Russell's viper phospholipase A2 and a designed peptide Leu-Ala-Ile-Tyr-Ser (LAIYS).
Chandra, V., Jasti, J., Kaur, P., Dey, S., Srinivasan, A., Betzel, C.h., Singh, T.P.(2002) Acta Crystallogr D Biol Crystallogr 58: 1813-1819
- PubMed: 12351825 Search on PubMed
- DOI: https://doi.org/10.1107/s0907444902013720
- Primary Citation Related Structures:
1JQ8 - PubMed Abstract:
Phospholipase A(2) (EC 3.1.1.4) is a key enzyme of the cascade mechanism involved in the production of proinflammatory compounds known as eicosanoids. The binding of phospholipase A(2) to membrane surfaces and the hydrolysis of phospholipids are thought to involve the formation of a hydrophobic channel into which a single substrate molecule diffuses before cleavage. In order to regulate the production of proinflammatory compounds, a specific peptide inhibitor of PLA(2), Leu-Ala-Ile-Tyr-Ser, has been designed. Phospholipase A(2) from Daboia russelli pulchella (DPLA(2)) and peptide Leu-Ala-Ile-Tyr-Ser (LAIYS) have been co-crystallized. The structure of the complex has been determined and refined to 2.0 A resolution. The structure contains two crystallographically independent molecules of DPLA(2), with one molecule of peptide specifically bound to one of them. The overall conformations of the two molecules are essentially similar except in three regions; namely, the calcium-binding loop including Trp31 (residues 25-34), the beta-wing consisting of two antiparallel beta-strands (residues 74-85) and the C-terminal region (residues 119-133). Of these, the most striking difference pertains to the orientation of Trp31 in the two molecules. The conformation of Trp31 in molecule A was suitable to allow the binding of peptide LAIYS, while that in molecule B prevented the entry of the ligand into the hydrophobic channel. The structure of the complex clearly showed that the OH group of Tyr of the inhibitor formed hydrogen bonds with both His48 N(delta1) and Asp49 O(delta1), while O(gamma)H of Ser was involved in a hydrogen bond with Trp31. Other peptide backbone atoms interact with protein through water molecules, while Leu, Ala and Ile form strong hydrophobic interactions with the residues of the hydrophobic channel.
- Department of Biophysics, All India Institute of Medical Sciences, New Delhi 110029, India.
Organizational Affiliation: