



RCSB PDB Help
Search and Browse > Advanced Search
Structure Motif Search
Introduction
What is a structure motif?
Structure motifs are the spatial or 3D arrangement of a small number of amino acids (at least 2) that have significance - e.g., form a catalytic or binding site. The amino acid residues making up the motif may be remote from one another in the 1D sequence or even be located in different polymer chains as long they are close to each other in 3D space (within 20 Å of each other). The structure motif search service (Bittrich et al., 2020) retrieves all occurrences of specific structure motifs in 3D structures available from RCSB.org.
The active site of the enolase superfamily is used as an example here (Figure 1, Meng et al., 2004). The enolase superfamily is a group of proteins diverse in sequence, yet largely similar in 3D structure that all catalyze removal of a proton from a carboxylic acid (Babbitt et al., 1996).
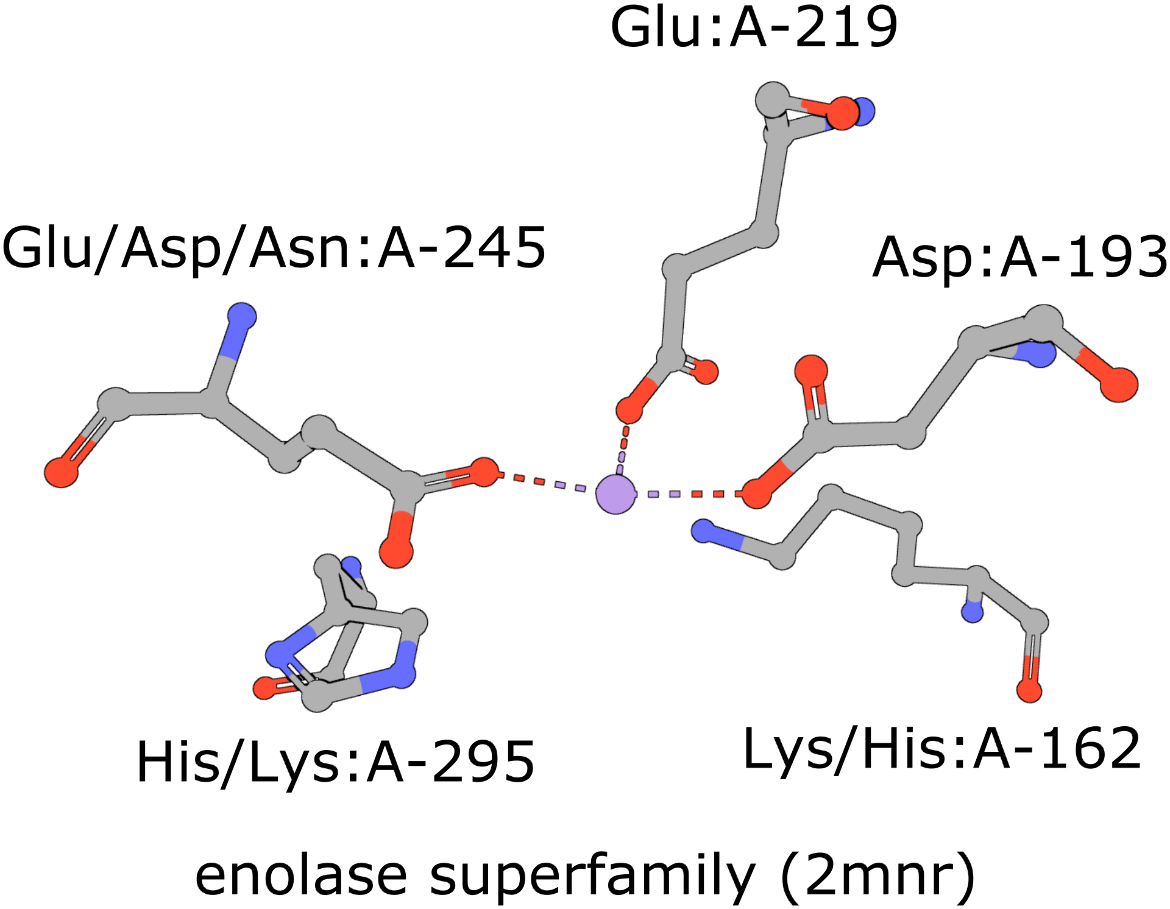
|
| Figure 1: Five residues representing the enolase superfamily are shown here. Note multiple amino acids are seen at three of these positions. The amino acids are identified by their amino acid name 3 letter abbreviation, chain ID (label_asym_id) and residue number (label_seq_id). |
When is this search useful?
The structure motif search service is particularly useful when you are interested in exploring the local structural properties of protein structures. This search service complements the structure search service and finds local, structural similarities between proteins. Search results only depend on the residues specified in the query, so it can identify local structural similarities even when the proteins have limited sequence or overall structural similarity. So, for example, this search can find similar ligand binding sites in unrelated proteins, regardless of whether the structures have a ligand bound in that neighborhood.
Detection of such structure motifs can provide valuable insights into the function(s) of previously uncharacterized proteins, especially ones that do not resemble other proteins at either the sequence or global structure level.
Documentation
The structure motif search service is accessible via the Mol* interface, where query residues (amino acids and nucleotides) can be specified in a visualized molecular structure, and the ‘Advanced Search’ panel, where the structure file identifier or location and query residue details can be specified by typing them into the interface.
Defining queries using Mol*
The RCSB Mol* plugin provides a convenient way to visualize a structure and define structure motif queries. The general Mol* documentation can be found here. Steps for specifying the structure motif query (Figure 2) are described here.
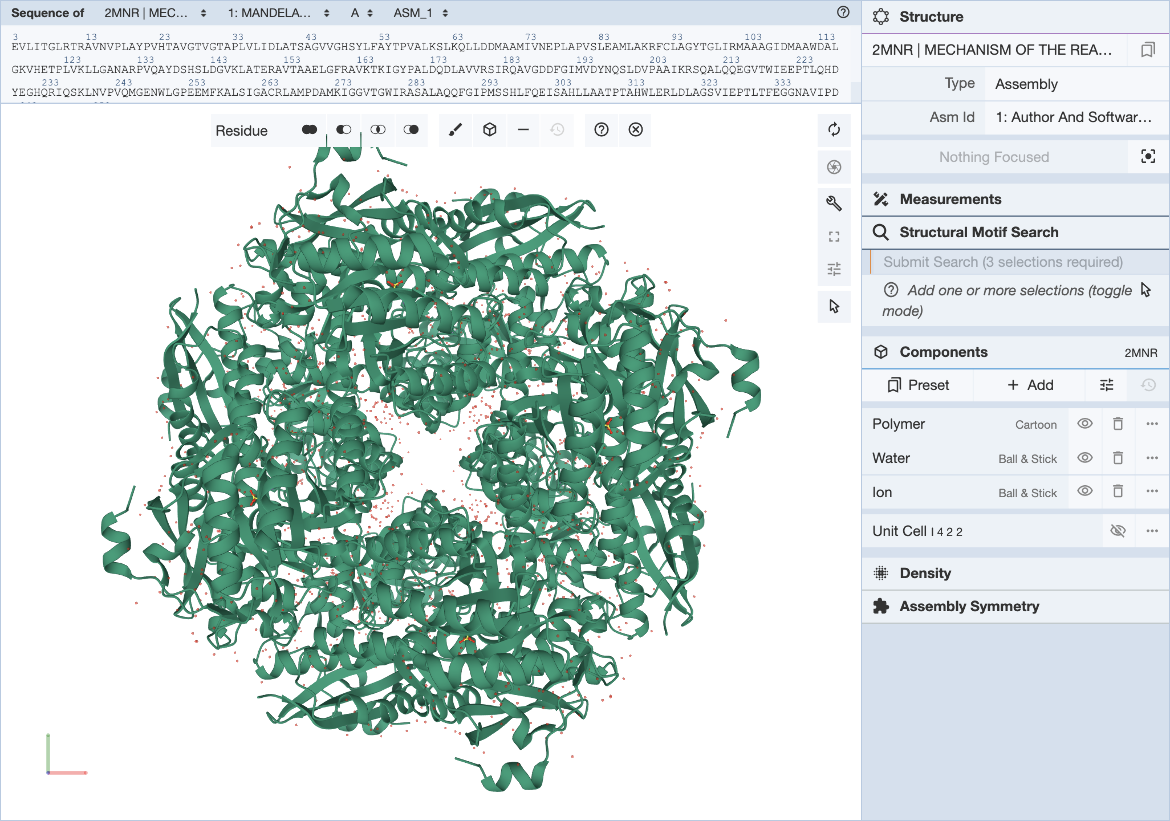
|
| Figure 2: Mol* user interface with Structure Motif Search panel expanded. |
To define a structure motif query for the enolase superfamily based on mandelate racemase (PDB ID 2mnr) and using the template described in (Meng, 2004) use the following steps.
In the Mol* interface, click and expand the ‘Structure Motif Search’ menu in the control panel on the right. Activate the selection mode of Mol* by clicking the mouse pointer icon and set the selection level to Residue (default). This allows you to select individual residues that will define the query (Figure 3).
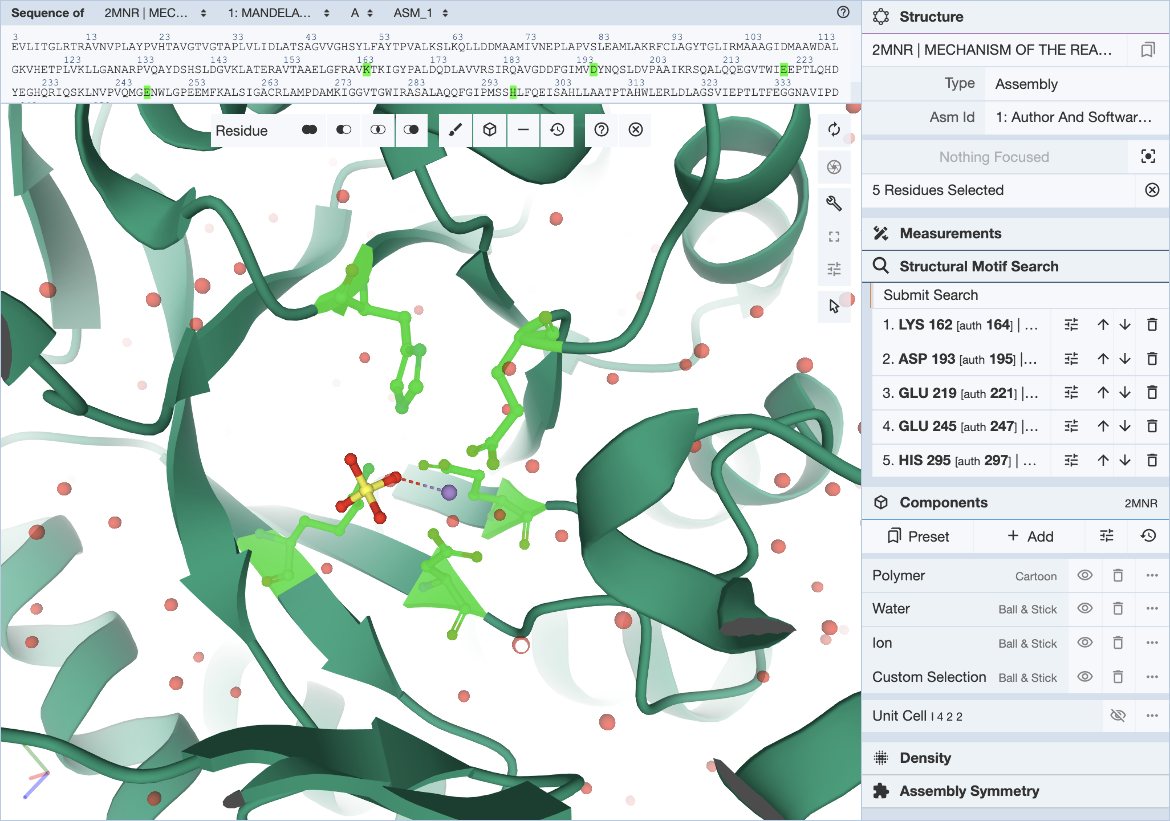
|
| Figure 3: Mol* user interface with motif selected. |
The 5 residues that constitute the template described in literature are used here to define the query motif.
Select individual residues by clicking on them either in the 3D canvas or in the sequence panel. The selected residues will be populated in the Structure Motif Search list in the control panel. Up to 10 residues may be included in this list. Add to the selection by clicking on additional residues, or remove residues by clicking on the trash icon in the residue list. The ‘Structure Motif Search’ element of Mol* behaves like the ‘Measurements’ panel.
Hover over the residue of interest to verify label_asym_id and label_seq_id. The information will appear in the tooltip in the bottom right corner of the Mol* panel. Author defined chain IDs and residue numbers will appear in square brackets if label and author identifiers are different. The sequence view at the top is particularly helpful when selecting residues by author numbering (Figure 4). Discrepancies between label_seq_id and auth_seq_id will be shown by Mol* in square brackets. Learn more about Identifiers in the PDB.
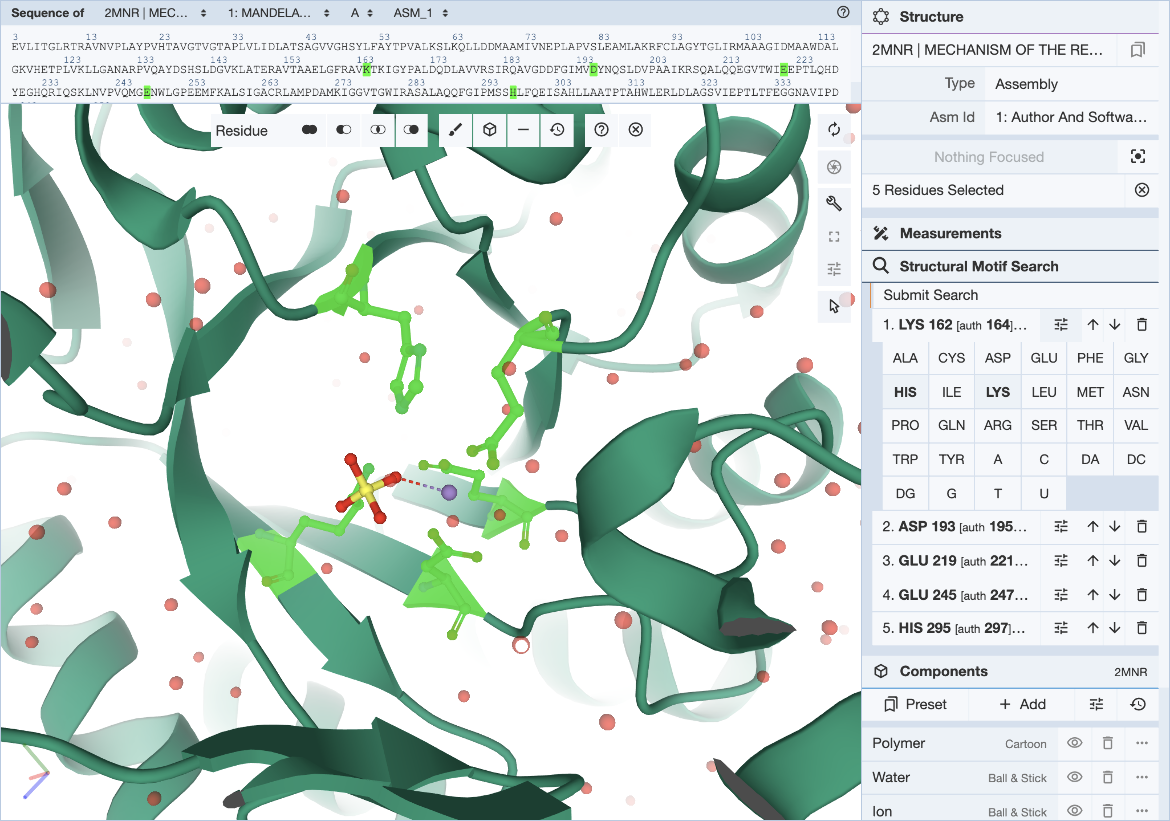
|
| Figure 4: Mol* user interface with exchange panel expanded and selections specified. |
In cases where a range of amino acids (or nucleotides) may realize the same biological function or bind the same ligand, it is possible to define position-specific exchanges in the query to accommodate possible variations in specific locations of the query structure motif.
For each entry of the residue list, exchanges can be specified individually by clicking on the options icon (three horizontal bars with short vertical lines intersecting them). This will open a panel with 20 amino acid and 8 nucleotide names. Click on all three-letter codes that should be considered as valid exchanges at the corresponding position. Only the original residue type is valid if no exchanges are defined. Make sure to include the original residue type when additional exchanges are defined. The number of exchanges per position is limited to 4.
Click the ‘Submit Search’ button. This will open a new browser tab and your query will be shown in the ‘Advanced Search’ panel.
Besides using the Mol* visualization options linked to Structure Summary pages of 3D structures available from the RCSB.org, a file upload functionality is available in the Mol* standalone tool (/3d-view). Once you upload a structure file from your local drive or by specifying a URL you can define a structure motif query as described above. Mol* will detect whether your file is an archive structure (and reference it using its Entry ID), a structure that was loaded using an external URL (and reference it using that link), or if you are visualizing a local file (in that case your file will be uploaded to our servers) and save the appropriate ID/link to the Advanced Search panel.
Defining queries using the 'Advanced Search' panel
The structure motif query can also be specified directly in the ‘Advanced Search’ panel. There are a few different options available to define the query.
Using the Structure ID for structures available from RCSB.org
To use this option you can enter a structure ID (PDB ID or RCSB.org assigned CSM ID) in the "Entry ID" box (Figure 5). A new button appears next to it called "Open in Mol*". Clicking on this button opens the structure in Mol* allowing you to select the query residues (as described above). Use options to select residues and the Mol* wizard to autofill the Structure Motif Search panel in the ‘Advanced Search’ panel. The panel also gives you opportunities to verify, refine, or extend your search manually.
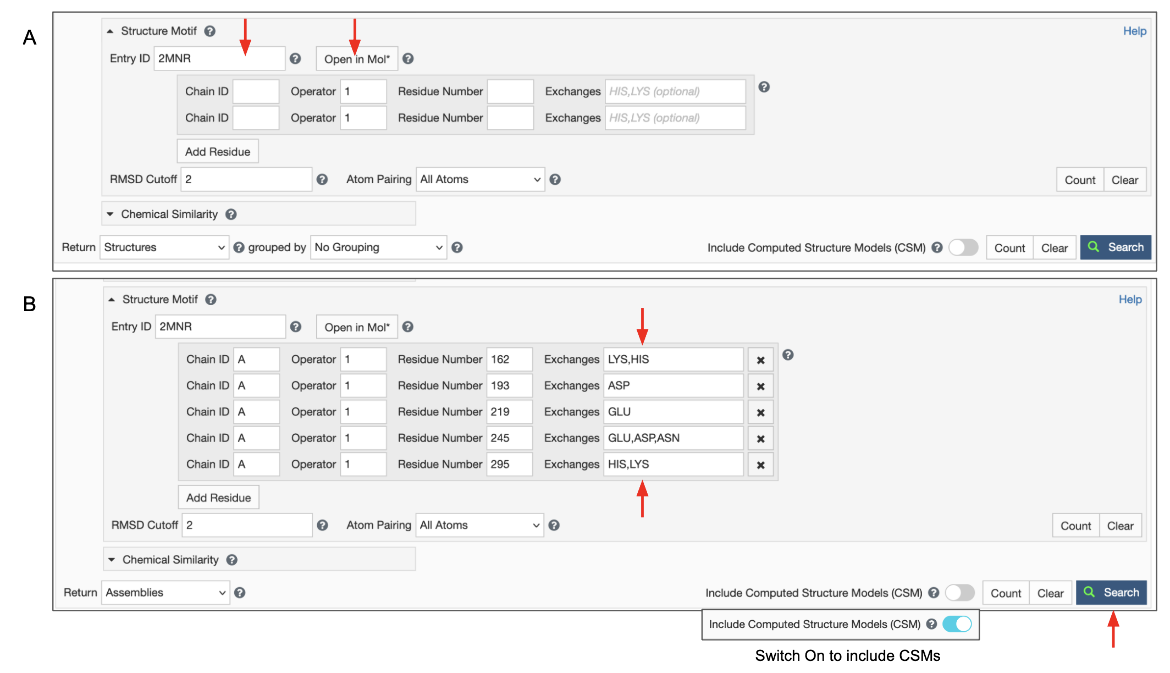
|
| Figure 5: Advanced Search panel showing the Structure Motif search panel. A. After typing in an Entry ID the "Open in Mol*" button appears (marked with red arrows). B. Structure motif search panel autofilled by Mol* and extended manually. |
Note: Clicking on the "Open in Mol*" button again (i.e., after the residue selections have been specified) opens the structure in Mol* and displays only the residues listed in the Structure Motif Search panel. Note: The To refine the selection of residues, turn on the selection more and select all the amino acids shown. Now display the polymer using options in the Controls panel and select additional (or fewer) residues to revise/refine the query and click on the Structure Motif Search >> Submit Search buttons. Note: if the Advanced search panel was manually extended to include alternate residues at a specific location, the "Open in Mol*" button will only display residues that are present in the structure. Possible alternatives listed in the panel will not be shown since those coordinates do not exist in the structure being visualized.
Using a File Link for structures that are available from other public data resources
This option can help find structures that are similar to a 3D structure available from public data resources e.g., from AlphaFold, RoseTTAFold, or ESMFold predictions but not included in RCSB.org.
To use this feature switch the input mode from “Entry ID” to “File Link” (Figure 6). Make sure to specify the URL as an “http” or “https” protocol. Specify the file format, which defaults to mmCIF, but BinaryCIF files are also supported. Specify the residues in your motif as usual. Decide on whether to include or exclude CSMs, and click on the blue Search button with a green magnifying lens icon to launch the search.
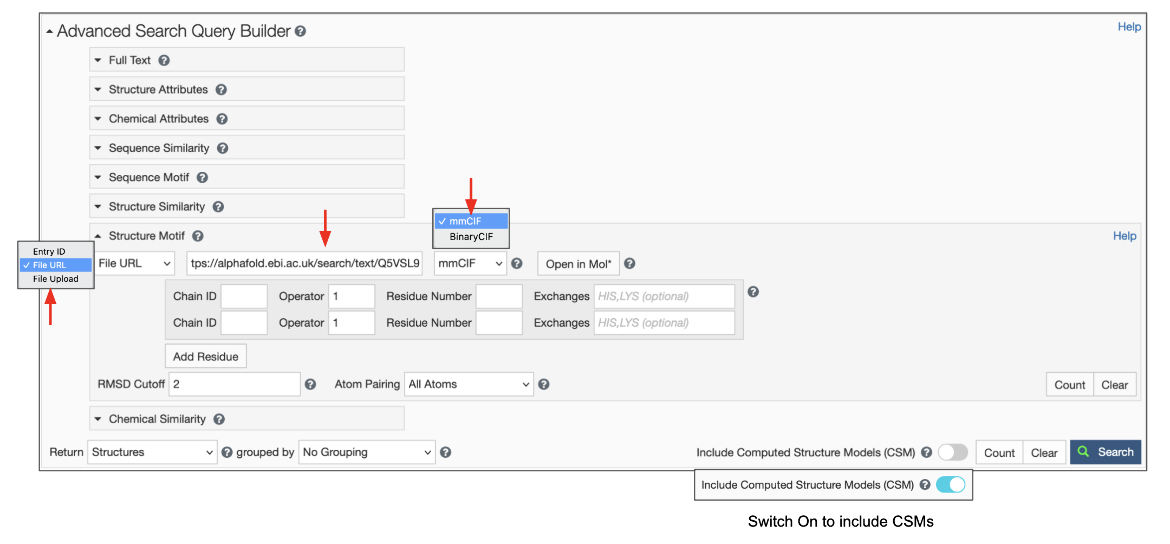
|
| Figure 6: Advanced Search panel showing the Structure Motif search panel for uploading a file using the File Link options. |
Uploading your own local file
This will give you access to a button that allows you to select a file from your file system. To use this feature, switch the input mode to “File Upload” (Figure 7). Files with “.cif”, “.bcif”, “.pdb”, and “.ent” extensions as well as their gzipped (“.gz”) variants are supported. Upon choosing the file, it will be automatically uploaded to our servers and the input mode will switch to “File Link”. Your file will be referenced by a unique URL. This random URL cannot be guessed by other users; however, please note that your data is accessible to anyone who knows this URL.
The uploaded file will be available for 90 days, which means you can bookmark your search or share it with colleagues for a limited amount of time. If you want to persistently reference a search in a publication, blog post or similar, you should upload your file to a file sharing service like Dropbox or Google Drive and reference it using the “File Link” feature. The same goes for queries that will be stored in MyPDB. Finally, the maximum file size supported is 10 MB, larger files also require the use of external file sharing services.
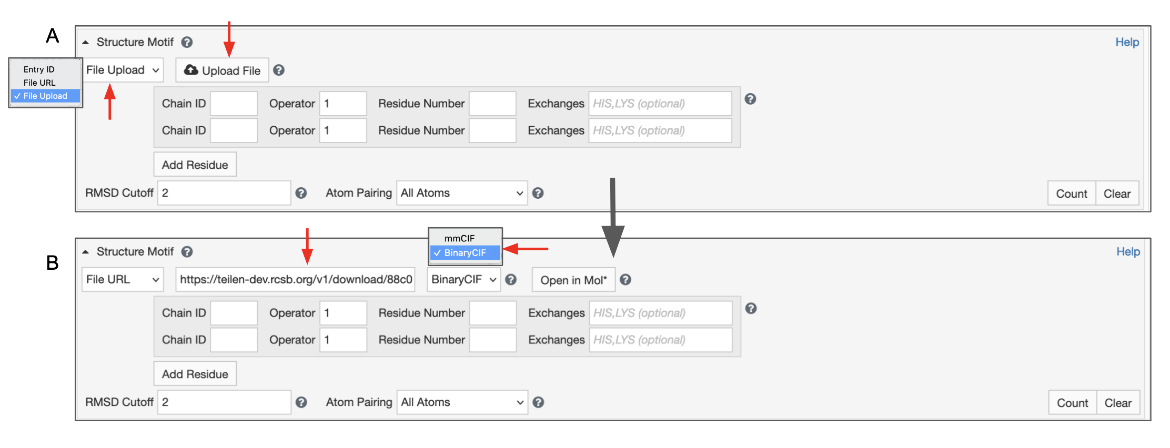
|
| Figure 7: Advanced Search panel showing the Structure Motif search panel for uploading a file from a local drive. A. Initial view B. After the file from the local drive is loaded to create a temporary web link. |
Regardless of the method used, once the structure coordinates are specified, a button “Open in Mol*” appears. Clicking on this button can be used to specify the residues in the structure motif. Clicking on this button again (i.e., after the residue selections have been specified) opens the structure in Mol* and displays only the residues listed in the Structure Motif Search panel.
Note:
- To refine the selection of residues, turn on the selection more and select all the amino acids shown. Now display the polymer using options in the Controls panel and select additional (or fewer) residues to revise/refine the query and click on the Structure Motif Search >> Submit Search buttons.
- If the Advanced search panel was manually extended to include alternate residues at a specific location, the "Open in Mol*" button will only display residues that are present in the structure. Possible alternatives listed in the panel will not be shown since those coordinates do not exist in the structure being visualized.
To fill in the structure motif residues manually, complete the following steps:
- Insert the PDB ID or RCSB.org assigned CSM ID that contains the query 3D structure motif.
- Specify 2 to 10 residues that make up the group of residues you want to find in other structures in the archive.
- The first box is for the polymer Chain ID (
label_asym_id) of the residues. Note that a motif may include amino acids from multiple polymer chains. - The Operator box is for optionally specifying the transformation operation that was used to generate a bioassembly (see PDB ID 2mnr as example). Identify operations by their
struct_oper_id. Combinations of operators are annotated like 1x61 or Px61. Set the value to '1' if you are referencing original coordinates. - The residue numbers included in the query are identified by their
label_seq_id. Note that in publications, residues are likely referenced by theirauth_seq_id, an identifier assigned by the authors. However, to define queries and report results the RCSB PDB website useslabel_seq_id. - Exchanges - Optionally, define position-specific exchanges or substitutions. Note, by default, only the residue type observed in the reference structure will be considered as valid. A set of comma-separated three-letter codes allows searching for different amino acids (or nucleotides) at the specified positions. Must include the original residue type to consider it at a particular position.
- Use the ‘Add Residue’ button to extend your selection to include additional amino acid residues in the structure motif, or use the ‘x’ button on the right to delete individual residues.
- The 'RMSD cutoff' parameter can be used to filter high RMSD hits that are unlikely to be biologically relevant.
- The 'Atom Pairing' parameter gives fine-grained control over the atom set that is considered for the alignment. By default all atoms are evaluated. Alternatively, only backbone, only side-chain, or only Cα/C4′ and Cβ/C1′ atoms can be selected for the RMSD calculation.
- Make sure to set the result type to ‘Assemblies’ to get detailed information on the result page that includes matched residue identifiers and reports the score of this hit. Note: A single entry may have more than one occurrence of the query structure motif. Since the motif may span more than one polymer chain, each occurrence is an assembly. If this option is not selected only PDB entries that contain the query motif are listed in the results.
Decide on whether to include or exclude CSMs, and click on the blue Search button with a green magnifying lens icon to launch the search.
How to interpret the result score?
The results are displayed as ‘Assemblies’.
All assemblies in the PDB archive that contain groups of residues that resemble the query motif are returned and sets of residues that match the query are identified by their label_asym_id and label_seq_id. Discrepancies between label_seq_id and auth_seq_id will be reported in square brackets. The label_comp_id of each residue is reported. The RMSD score of the match is provided as well (see Figure 8).
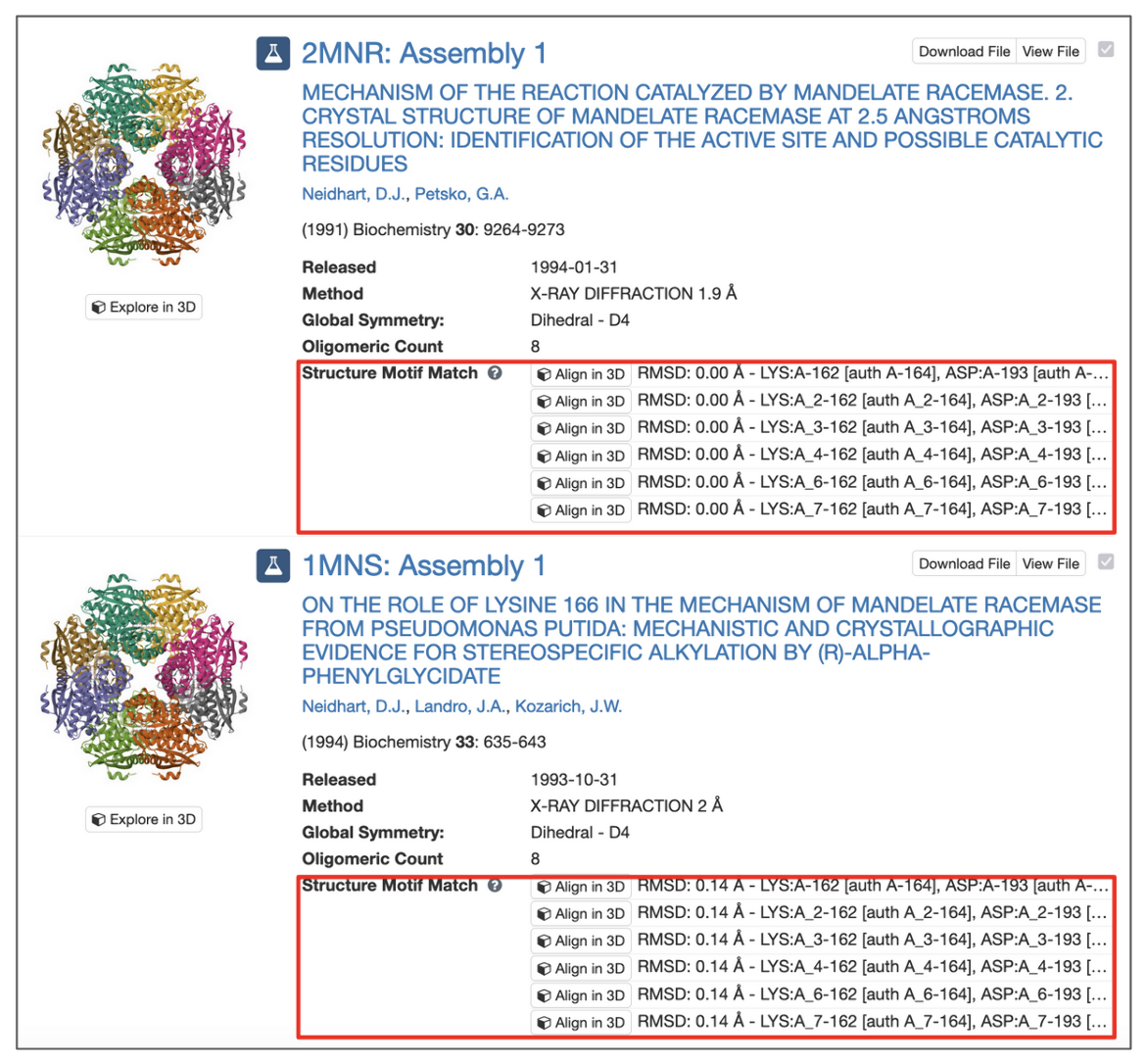
|
| Figure 8: Structure Motif Search results with match context. |
All potential matches are reported with a root-mean-square deviation (RMSD) score, which is computed by aligning each identified match to the query motif and measuring the displacement of each matched atom. Values of 0.0 Å indicate optimal alignment, higher values occur for dissimilar groups of residues.
Motifs may occur in symmetry partners of the deposited coordinates. In these cases, chain identifiers will include the corresponding struct_oper_id after an underscore (e.g., LYS:A_2-162).
The 'Align' button at the beginning of each line launches a Mol* view that shows the superposition of query motif and selected match.
Limitations of Structure Motif Search
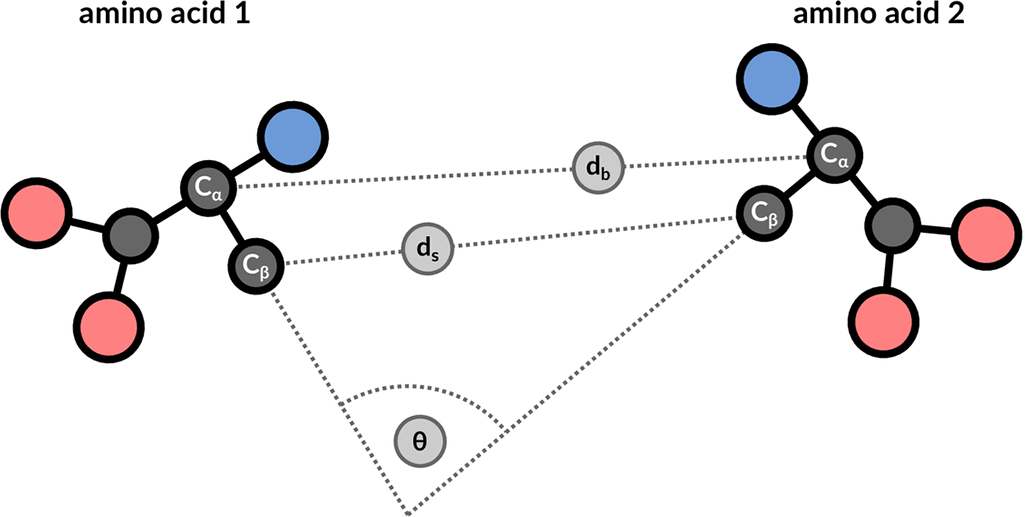
The structure motif search service is a heuristic search with a false negative rate <2%. This means that 1 in 50 relevant hits will get missed when compared to a much slower exhaustive search strategy. The service uses 3 features to describe the geometric properties of all residue pairs present in the query motif (Figure 9): backbone distance (db), side-chain distance (ds), and the angle θ between the CαCβ vector of both amino acids. Hits will get missed if one of these properties differs too much. Tolerance values are 1 Å for distances and 20° for the angle property.
The false positive rate for hits with low RMSD values <0.5 Å tends to be 0, but the false positive rate increases for hits with higher RMSD values. This also means that no hits will be found in structures that contain only a Cα trace.
|
| Figure 9: 3 geometric properties are used to describe residue pairs: backbone distance between Cα atoms, side-chain distance between Cβ atoms, and angle between the corresponding vectors. |
Details about the search algorithm and scoring are discussed in Bittrich et al., 2020. In particular, see Figure 3 and the accompanying discussion of observed false negatives. The 'For advanced users' section provides information on how to run structure motif queries with increased tolerance values that lower false negative rates at the expense of higher runtimes.
Examples
The structure motif search service finds resemblances of 2 to 10 residues that are in spatial proximity. Interesting motifs are defined in literature and available in resources such as the Catalytic Site Atlas (CSA). It is applicable for a number of example queries. All given identifiers are label_asym_id and label_seq_id.
Table 1: Examples of Structure Motif Search
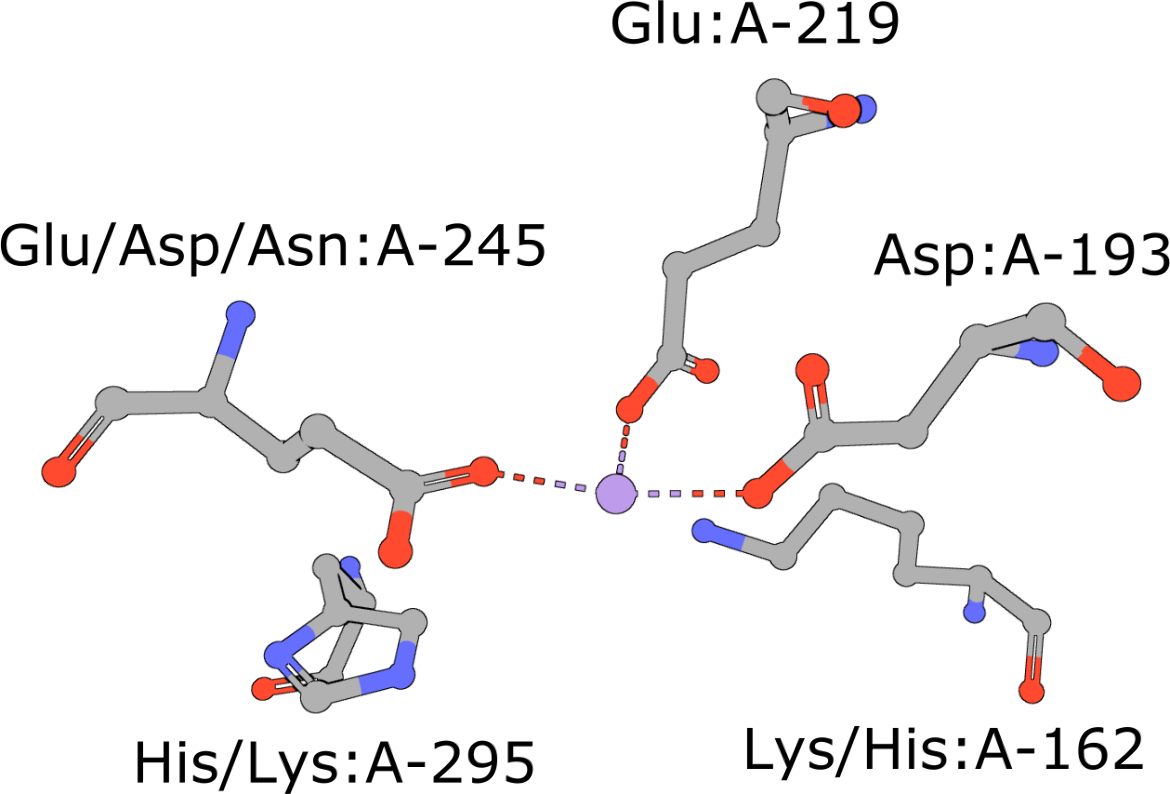 |
Template of the enolase superfamily
(execute query) The enolase superfamily is a group of proteins diverse in sequence, yet largely similar in 3D structure that all catalyze removal of a proton from a carboxylic acid (Babbitt, 1996). The structure motif supporting this catalytic function (Meng, 2004) is represented in PDB ID 2mnr. |
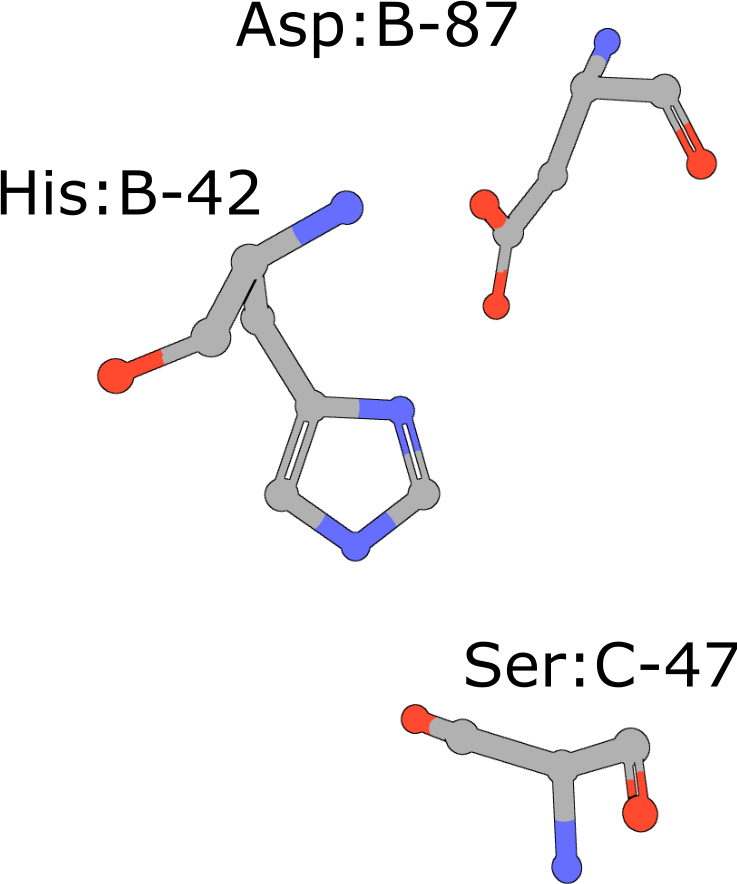 |
Catalytic triad of serine proteases
(execute query) Many hydrolases use a serine nucleophile during catalysis. Canonical serine protease catalytic triads are composed of His, Asp, and Ser residues (PDB ID 4cha). Typically these residues occur within two polypeptide chains, because many of these proteases are initially made as zymogens that require activation by proteolytic processing (Hedstrom, 2002) to prevent uncontrolled digestion of proteins within the cell. You can also combine your query with keywords to narrow the result set and find more interesting occurrences of the query motif. |
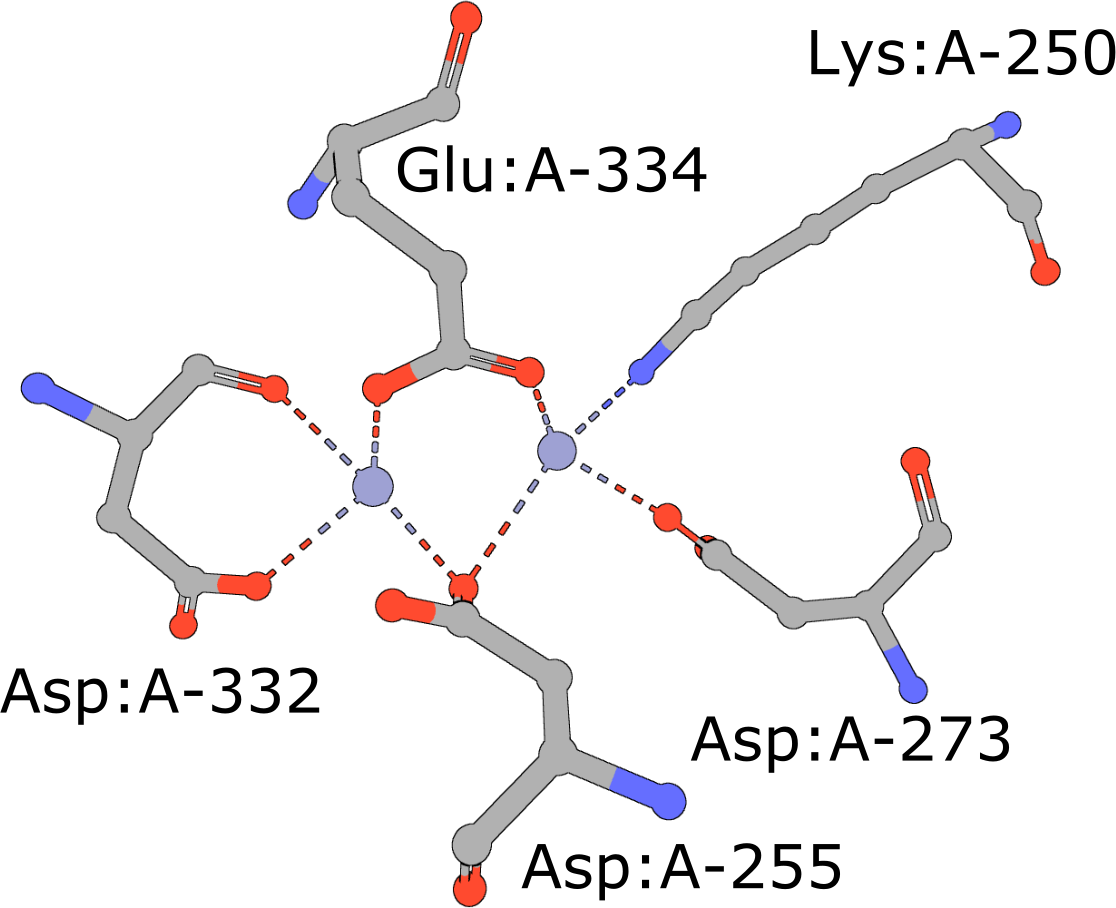 |
Aminopeptidase
(execute query) Aminopeptidases play important roles in protein degradation by removing residues from the N- or amino terminus of polypeptide chains (Burley, 1990). Bovine leucine aminopeptidase (BLLAP) is a homohexameric enzyme with 32 quaternary symmetry. The active site of BLLAP contains two adjacent zinc ions separated by ∼2.9 Å and coordinated by the sidechains of five conserved residues Lys, Asp, Asp, Asp, and Glu (PDB ID 1lap). |
 |
Zinc Finger
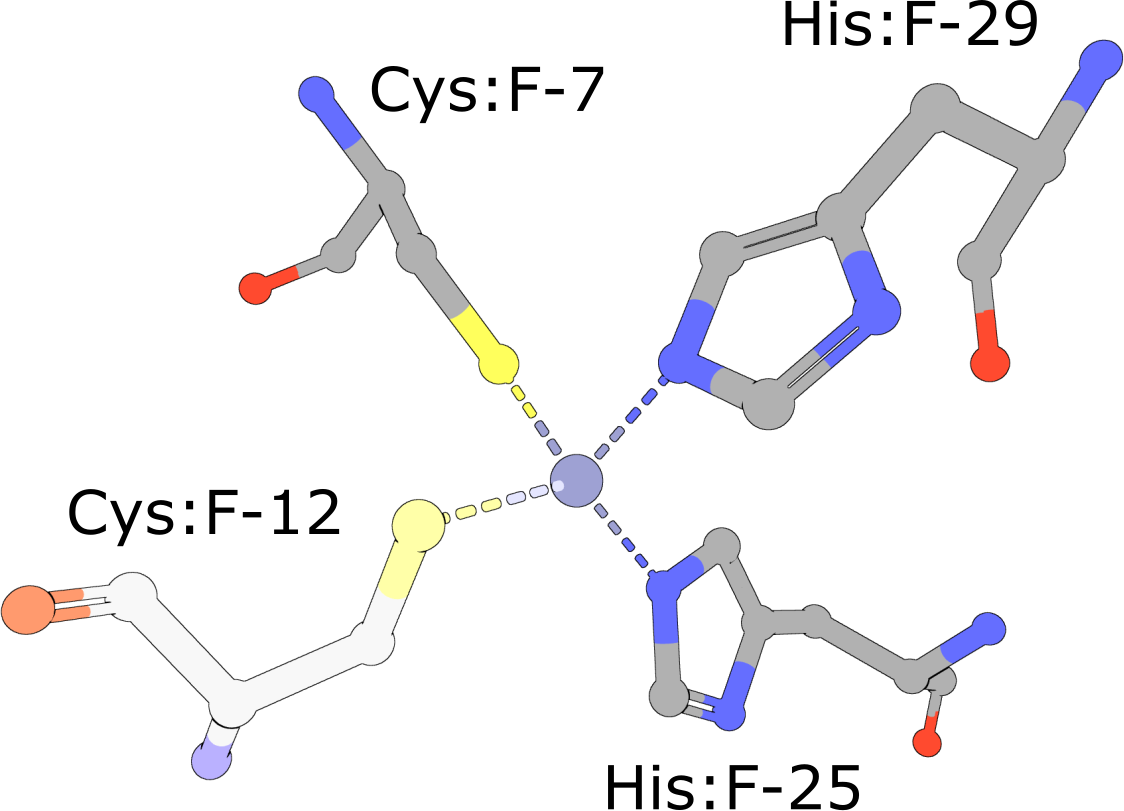
(execute query) Eukaryotic transcription factors often contain His2/Cys2 Zinc Finger domains (PDB ID 1g2f) that bind DNA. These motifs are composed of two cysteine and two histidine residues, which stabilize a small ββα domain structure that envelopes and coordinates a single zinc ion (Pabo, 2001). In the absence of the zinc ion, these domains do not adopt compact, folded structures and are incapable of binding DNA. |
 |
RNA G-tetrad
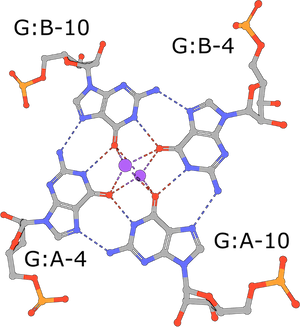
(execute query) G-tetrads are a common nucleic acid association motif (PDB ID 3mij). They are composed of guanines and stabilized by Hoogsteen base pairings. The four O6 oxygen atoms coordinate monovalent ions, such as K+, and individual tetrads tend to be stacked one atop the other (Burge, 2006). |
 |
Cadmium Coordination
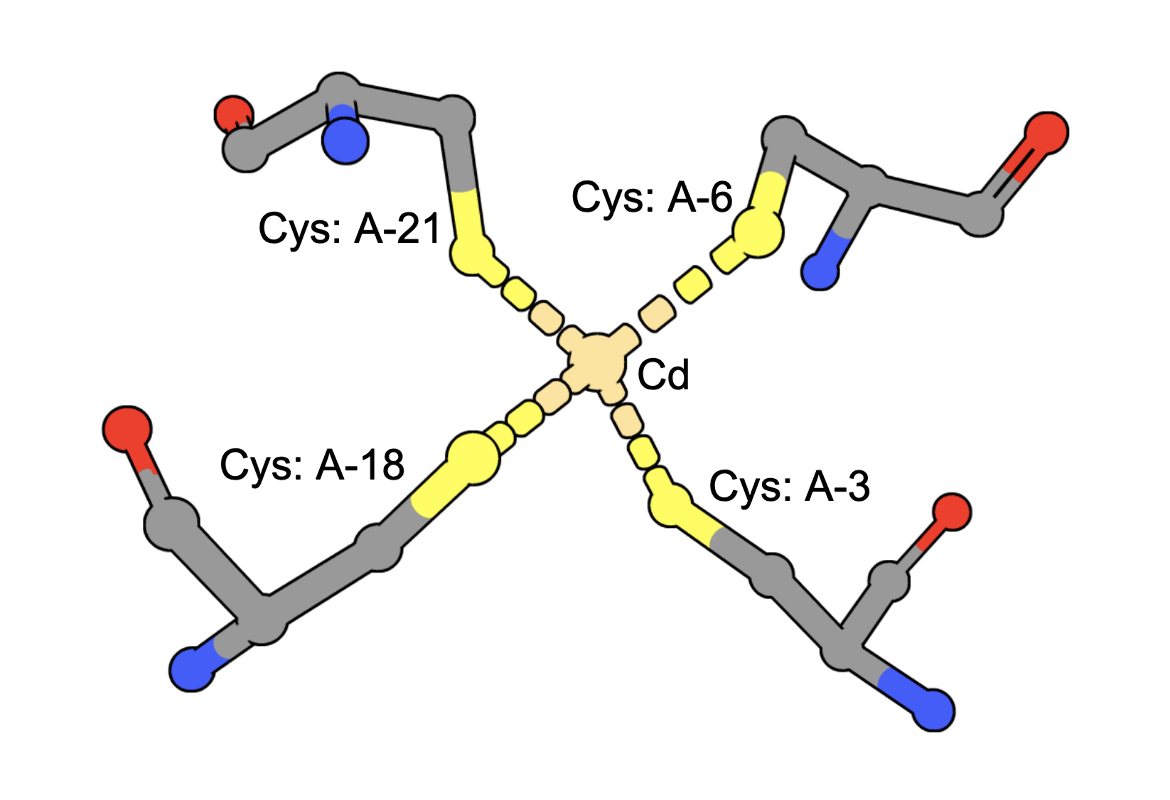
(execute query) Cadmium ions can bind to sulfur containing amino acids (e.g., Cys) in proteins. A query to find structures with Cd bound by four Cys residues can be constructed by combining two types of queries: a. structure motif search - for structures with 4 Cys residues around an ion (often Zinc is found in these geometries), AND b. chemical attribute search - for structures that contain a Cd ion The query finds an intersection of (structures with Cd) and (structures with 4 Cys residues positioned to coordinate an ion). An example from the results shows a structure that has a Cd bound to four Cys amino acids from the PDB ID 5sbj. Note: It is possible that structures have the Cd but it is not coordinated by the 4 Cys residues. Thus the results of this query should be examined carefully to ensure that they include at least one cadmium coordinated by Cys4. |
For advanced users
All Java source-code is publicly available on GitHub (github.com/rcsb/strucmotif-search), and the project is distributed as a Maven artifact.
We encourage interested users to set up a local installation of the structure motif search service. This allows you to configure the tool for your exact requirements and gives fine-grained control over all parameters, some of which are not exposed on rcsb.org. Additional features include:
- Increased tolerance values that allow one to retrieve more dissimilar hits
- Definition of query motifs using custom structures that are not part of the PDB archive (such as AlphaFold structures)
- Screening for occurrences of known motifs in a structure of unknown function
References
- Bittrich S, Burley SK, Rose AS (2020) Real-time structural motif searching in proteins using an inverted index strategy. PLoS computational biology. 16(12): e1008502, doi: 10.1371/journal.pcbi.1008502
- Meng EC, Polacco BJ, Babbitt PC (2004) Superfamily active site templates. PROTEINS: Structure, Function, and Bioinformatics. 55(4): 962–976, doi: 10.1002/prot.20099.
- Babbitt PC, Hasson MS, Wedekind JE, Palmer DR, Barrett WC, Reed GH, et al. (1996) The enolase superfamily: a general strategy for enzyme-catalyzed abstraction of the α-protons of carboxylic acids. Biochemistry. 35(51): 16489–16501, doi: 10.1021/bi9616413.
- Hedstrom L. (2002) Serine protease mechanism and specificity. Chemical reviews. 102(12): 4501–4524, doi: 10.1021/cr000033x.
- Burley SK, David PR, Taylor A, Lipscomb WN (1990) Molecular structure of leucine aminopeptidase at 2.7-A resolution. Proceedings of the National Academy of Sciences. 87(17): 6878–6882.
- Pabo CO, Peisach E, Grant RA (2001) Design and selection of novel Cys2His2 zinc finger proteins. Annual review of biochemistry. 70(1):313–340, doi: 10.1146/annurev.biochem.70.1.313.
- Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S (2006) Quadruplex DNA: sequence, topology and structure. Nucleic acids research. 34(19): 5402–5415, doi: 10.1093/nar/gkl655.


